Conheça mais sobre nossas soluções
BDASeq®
Descubra biomarcadores e alvos para drug design com acurácia
Por analisar de forma quantitativa e simultânea a expressão gênica global de células e/ou tecidos de interesse, o sequenciamento de RNA (RNA-Seq) se tornou uma valiosa e indispensável técnica de busca de biomarcadores e/ou alvos terapêuticos (druggable genes).

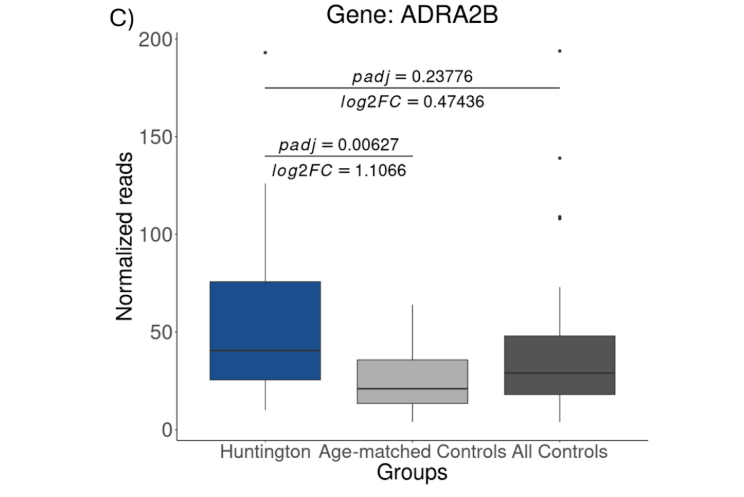
No entanto, a análise dos dados de RNA-Seq pode ser uma tarefa laboriosa, já que exige diferentes pacotes e linguagens de programação, tornando a análise pouco acessível para profissionais não habituados com bioinformática. Além disso, a multitude de ferramentas disponíveis para analisar os genes diferencialmente expressos, somada às vantagens e desvantagens oferecidas por cada uma destas ferramentas, pode exigir estudos prévios para identificar a melhor estratégia analítica a ser empregada. Uma vez analisada a expressão diferencial, os resultados gerados fornecem uma lista de genes diferencialmente expressos (GDEs), que pode chegar até milhares de GDEs. Com isso, é necessário que o investigador adote critérios que permitam identificar dentre os GDEs identificados aqueles que possam servir como potenciais biomarcadores ou alvos terapêuticos (druggable genes). Entretanto, por vezes, os critérios adotados podem levar a resultados falso-positivos, resultando na identificação de genes que embora possam apresentar diferenças estatísticas significativas entre casos e controles (FDR < 0,05) e valores significativos de log2 fold-change (+1,0 < log2FC < -1,0), podem não servir como biomarcadores, como mostrado no caso real descrito na Figura 1.
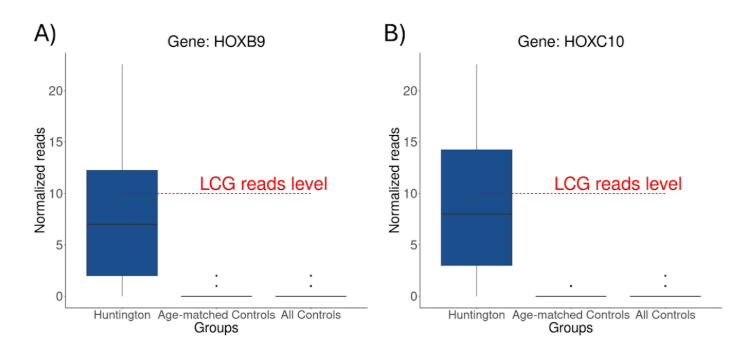
Exemplo: Buscando identificar biomarcadores para a doença neurodegenerativa de Huntington (DH), Labadorf et al. (2015; link para https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0143563 ) comparou expressão de mRNAs do córtex pré-frontal de 20 indivíduos portadores da doença e 49 indivíduos neurologicamente saudáveis (não portadores da DH). Usando as técnicas convencionais de análise de RNA-Seq, os autores identificaram 5.480 genes diferencialmente expressos no córtex pré-frontal dos indivíduos portadores da DH. Com base na ordenação dos valores de log2FC, os autores identificaram 39 HOX genes como potenciais biomarcadores. No entanto, ao analisar as medianas das reads normalizadas destes genes nos indivíduos portadores da DH, fica evidente que estas se encontram abaixo de 10, valores este reconhecido como limite inferior para considerar um gene como expresso, como mostrados nos exemplos das análises dos genes HOXB9 (A) e HOXC10 (B). No entanto, por não serem expressos nos indivíduos controle, os valores de log2FC gerados levam a resultados falso-positivos que impactam negativamente na identificação de biomarcadores.
Por esta razão, o BDASeq® reúne todas os pacotes necessários para analisar os dados transcriptômicos. Dividido em três camadas (processamento, análise de expressão diferencial e busca de alvos), o BDASeq® permite fazer o upload dos dados de RNA-Seq diretamente do computador ou do repositório público do Sequence Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra ), permitindo aumentar o número amostral ou até mesmo buscar alvos sem a necessidade de coletar e sequenciar amostras biológicas. Além disso, a camada de processamento do BDASeq® conta com o algoritmo Propensity Match Score (PMS), que auxilia na seleção apropriada de amostras controle com base na distribuição de variáveis de relevância, garantindo resultados mais acurados, conforme demonstrado por Dias-Pinto et al. (2024 e 2025).
A camada de processamento também reúne todas as ferramentas necessárias à análise dos dados de acordo com as Boas Práticas da análise de RNA-Seq, realizando a verificação da qualidade dos sequenciamentos, o alinhamento/mapeamento das reads com o genoma de referência, e a contagem das reads que serve com input para a camada de análise da expressão diferencial.
Na camada de análise da expressão diferencial, o BDASeq® conta com oito diferentes ferramentas que analisam individualmente a expressão diferencial. Usando o exclusivo algoritmo Recursive Method Combination (RMC), os resultados são combinados de formada inteligente, reduzindo os erros de tipo I e II de cada ferramenta, aumentando a acurácia de identificação dos GDEs.
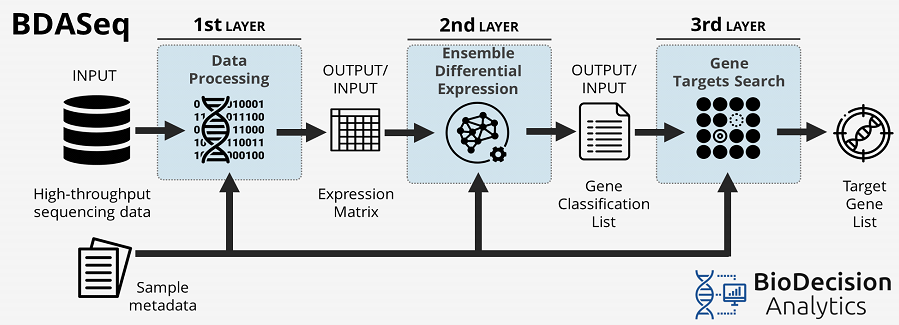
Por fim, todos os transcritos anotados no genoma de referência são classificados, conforme a classificação desenvolvida pela BioDecision (Tabela 1, Figura 2). Com isso, é possível verificar o status de cada gene individualmente.
Tabela 1. Classificação dos genes quanto ao seu nível de expressão em relação ao grupo controle
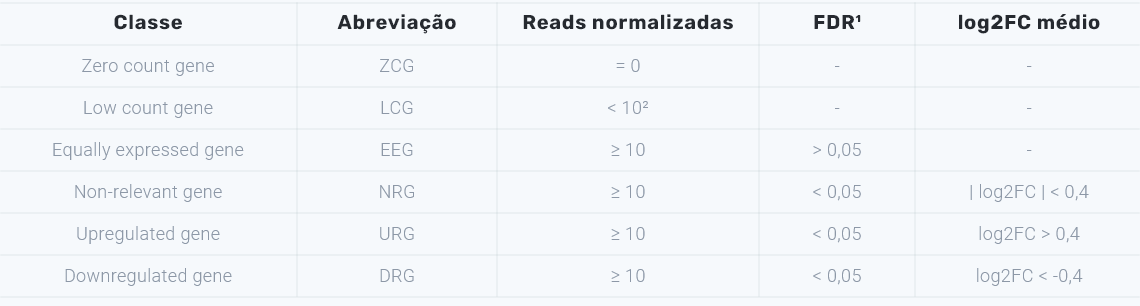
¹FDR – false-discovery rate ou p-valor ajustado máximo entre todos os oito métodos.
²um gene é considerado expresso quando possuir valores de reads normalizadas maiores que 10 em menos 70% das amostras que compreendem a volumetria do menor grupo (caso ou controle).
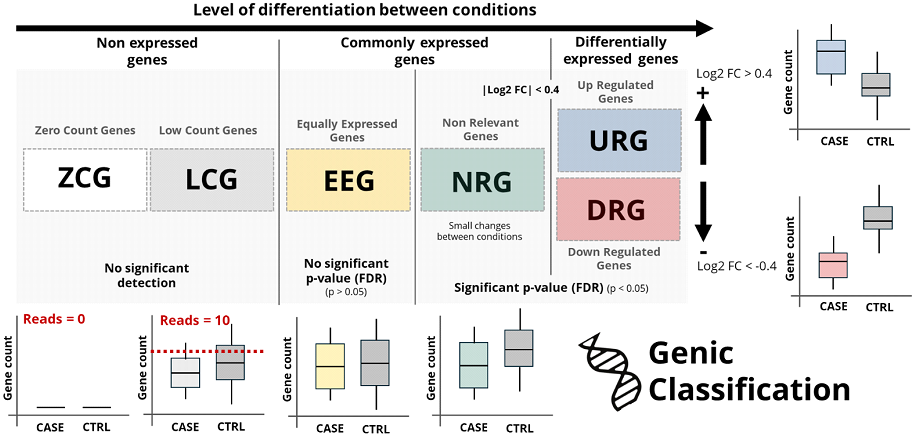
Figura 2. Sistema de classificação gênica exclusivo que permite classificar todos os transcritos expressos pelo genoma da espécie analisada como: zero count genes (ZCG) – genes com valores de reads igual a zero; low count genes (LCG) – genes com valores de reads abaixo de 10; equally expressed genes (EEG) – genes que se expressam de forma estatisticamente semelhantes entre casos e controles, ou seja possuem FDR > 0,05; non-relevant genes (NRG) – genes que embora apresentem diferenças estatísticas significativas entre casos e controles (FDR < 0,05), estas diferenças não são biologicamente relevantes (|log2FC| < 0,04); upregulated genes (URG) – genes positivamente regulados, i.e., genes cuja expressão se encontra aumentada nos casos em relação aos controle (FDR < 0,05 e log2FC > 0,04); e downregulated genes (DRG) – genes negativamente regulados, i.e., genes cuja expressão se encontra reduzida nos casos em relação aos controle (FDR < 0,05 e log2FC > 0,04). Desta forma, apenas os URG e DRGs são reconhecidos como diferencialmente expressos. Imagem de propriedade intelectual da BioDecision Analytics℠.
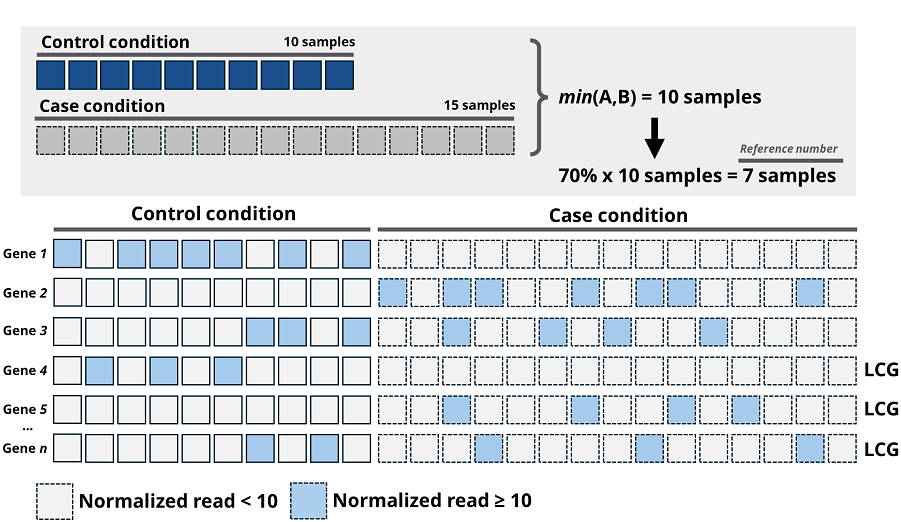
Figura 3. Critério de classificação dos LCGs adotado pelo BDASeq. Modelo classifica como LCGs genes que apresentam valores de contagem normalizada menor que 10 (para os métodos DESeq2, EBSeq, dearseq, Wilcoxon e regressão logística de Firth) ou 0,3 (para os métodos edgeR, limma-voom e NOISeq), em mais de 70% da volumetria de referência, que é calculada pela volumetria total da categoria (caso ou controle) com o menor número amostral. Imagem de propriedade intelectual da BioDecision Analytics LTDA.
Diferente de qualquer outro método disponível, o BDASeq® conta com algoritmos de inteligência artificial e estatística extensivamente validados que permitem buscar dentre os GDEs, potenciais candidatos a biomarcadores e/ou druggable genes.
Para conhecer mais sobre o BDASeq®,
entre em contato conosco.